

Scientists from the Institut Pasteur and CNRS, in collaboration with scientists from the Institut Gustave Roussy and CEA, have succeeded in restoring normal activity in cells isolated from patients with the premature aging disease Cockayne syndrome. They have uncovered the role played in these cells by an enzyme, the HTRA3 protease. This enzyme is overexpressed in Cockayne syndrome patient cells, and leads to mitochondrial defects, which in turn play a crucial role in the appearance of symptoms leading to aging in affected children. These findings, published in the journal PNAS Plus, describe one of the hitherto unknown mechanisms responsible for premature aging. They could also shed light on the normal aging process.
Press release
Paris, May 18, 2015
Rare genetic diseases cause accelerated premature aging. To date, there is no treatment for these pathologies. Understanding the causes of premature aging diseases may also help elucidating the process of normal aging. One such disease, Cockayne syndrome (CS), has an incidence of about 2.5 per million births and, in its most severe form, is associated with a life span of less than seven years. Children with Cockayne syndrome show marked signs of premature aging, such as loss of weight, hair, hearing and sight, as well as facial deformation and neurodegeneration.
Cockayne syndrome is caused by mutations in either of two genes involved in the repair of DNA damage induced by ultraviolet (UV) rays. CS patients are hypersensitive to sunlight and burn easily. For decades it was believed that the premature aging process associated with this disease was essentially caused by DNA repair deficiency.
By comparing cells from CS patients and from patients with another, related syndrome causing only UV hypersensitivity, the team led by Miria Ricchetti (Institut Pasteur) with Laurent Chatre (CNRS, at the Institut Pasteur), in collaboration with Alain Sarasin (CNRS, at the Institut Gustave Roussy) and Denis Biard (CEA), discovered that the defects in CS cells are actually due to excessive production of a protease (HTRA3), and induced by oxidative cell stress. In CS cells, HTRA3 degrades a key component of the machinery responsible for DNA replication in mitochondria (the cellular “powerhouses”), thereby affecting mitochondrial activity.
Until now, neurodegeneration and aging have largely been attributed to the damage inflicted on cells by mitochondrial free radicals. The new study shows that free radicals also activate the expression of a protein known as HTRA3, which is particularly damaging to mitochondria. This onslaught on the mitochondrial core is a key factor in the degeneration of cells in patients suffering from premature aging.
By means of two new therapeutic strategies, using an HTRA3 inhibitor or a broad-spectrum antioxidant to capture free radicals, the scientists succeeded in restoring normal levels of this protease. They were accordingly able to restore mitochondrial function in CS patients. This progress both paves the way for new therapeutic approaches, which could soon be tested in patients, and provides new diagnostic tools.
The Institut Pasteur, CNRS, CEA and IGR have filed a patent application for methods of diagnosing and treating premature aging using the protease HTRA3.
These defective mechanisms may also occur, but at a slower rate, in healthy cells, leading to physiological aging. The development of therapeutic strategies targeting premature aging diseases could therefore open new research possibilities in terms of preventive therapies for the pathologies associated with normal aging.
This research was supported by the Institut Pasteur, the French National Research Agency (ANR) and the French Muscular Dystrophy Association (AFM).
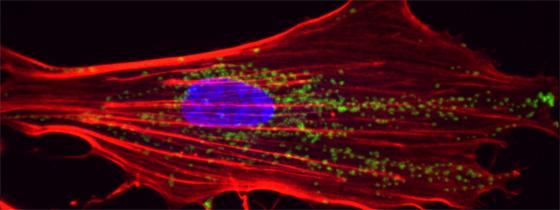
Illustration: 3D reconstruction of a primary fibroblast from the skin of a patient with Cockayne syndrome: mitochondria are shown in green, the nucleus in blue and the actin network (the cell’s “skeleton") in red. © Institut Pasteur
Source
Reversal of mitochondrial defects with CSB-dependent serine protease inhibitors in patient cells of the progeroid Cockayne syndrome, PNAS Plus, May 18, 2015
Laurent Chatre1,2,, Denis Biard3, Alain Sarasin4,5 and Miria Ricchetti1,2,6
1Institut Pasteur, Department of Genomes and Genetics, Unité de Génétique Moléculaire des Levures, 25 Rue du Dr. Roux, 75724, Paris, France,
2CNRS UMR 3525, Team Stability of Nuclear and Mitochondrial DNA,
3CEA, DSV, IMETI, SEPIA, Team Cellular Engineering and Human Syndromes, F-92265 Fontenay-aux-Roses, France,
4UMR8200 CNRS Laboratory of Genetic Stability and Oncogenesis, Institut de cancérologie Gustave Roussy, and University Paris-Sud, 114 rue Edouard Vaillant, 94805 Villejuif, France,
5Department of Genetics, Institut de Cancérologie Gustave Roussy.
6Corresponding author.