

Des chercheurs de l’Institut Pasteur, de l’Institut de la Vision, de l’Inserm et de l’Université Pierre et Marie Curie ont élucidé l’origine de la cécité qui survient dans le syndrome de Usher de type I (cause la plus fréquente de surdité-cécité chez l’homme). Les scientifiques ont également démontré pourquoi le rongeur, seul modèle animal disponible aujourd’hui pour cette pathologie, n’est pas atteint par la cécité observée chez l’homme. Ces travaux impliquent l’orientation des futures recherches vers la production d’un modèle animal chez le primate. Ce dernier permettra ensuite de progresser vers une approche thérapeutique de la cécité chez les patients atteints du syndrome de Usher de type I. Ces recherches font l’objet d’une publication le 8 octobre dans le Journal of Cell Biology.
Communiqué de presse
Paris, le 9 octobre 20112
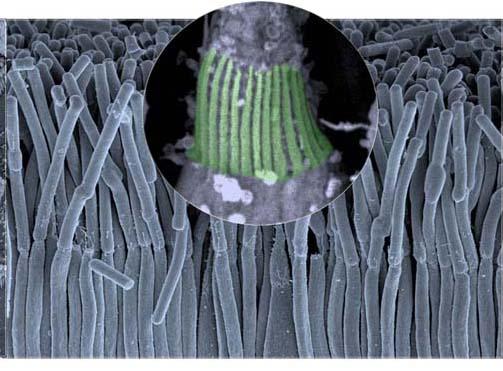
Les travaux du Pr Christine Petit , chef de l’Unité de recherche « Génétique et physiologie de l'audition » de l’Institut Pasteur, en collaboration avec le Dr Aziz El-Amraoui (Institut Pasteur) et le Pr José-Alain Sahel (Institut de la Vision), relancent l’espoir : les chercheurs viennent de découvrir l’origine de la rétinite pigmentaire chez les patients atteints du syndrome de Usher de type I. Il s’agit d’un défaut dans l’organisation d’édifices cellulaires indispensables au maintien de la vision, les processus caliciels. Ce défaut est causé par le dysfonctionnement d’une ou de plusieurs protéines, identifiées au nombre de 5 par les chercheurs et qui assurent la cohésion des processus caliciels. La structure des processus caliciels a pu être observée en haute résolution grâce à l’utilisation de techniques de microscopie électronique (cf photographie).
Dans leur étude, les chercheurs ont également mis en évidence une différence majeure entre les cellules photoréceptrices des rongeurs et celles des primates. Contrairement aux seconds, les premiers ne possèdent pas de processus caliciels. C’est la raison pour laquelle on n’observe pas de rétinite pigmentaire lorsque les gènes qui sont responsables du syndrome de Usher chez l’homme sont mutés chez la souris.
Pionnière dans l’étude des surdités héréditaires, l’équipe du Pr Christine Petit avait déjà établi la responsabilité de ces 5 mêmes protéines dans la survenue des troubles de l’audition du syndrome de Usher de type I. Ces recherches avaient notamment permis d’ouvrir la voie à la connaissance des bases moléculaires de l’audition. Avec ces nouveaux résultats, les chercheurs espèrent mettre au point un meilleur modèle animal, et ainsi être en mesure de développer rapidement une approche thérapeutique de la rétinite pigmentaire pour le syndrome de Usher.
Ces travaux ont notamment bénéficié du soutien de la communauté européenne HEALTH-F2-2010-242013 (TREATRUSH), LHW-Stiftung, Fondation Raymonde & Guy Strittmatter, FAUN Stiftung (Suchert Foundation), Conny Maeva Charitable Foundation, Fondation Orange, l’Agence Nationale de la Recherche [ANR-10-LABX-65], [ANR-07-MRARE-009-01], "the Foundation Fighting Blindness Paris Center Grant" and the Fondation Voir et Entendre.
--
Illustration – Copyright Institut Pasteur / Cataldo Schietroma et Vincent Michel
Légende - la structure des processus caliciels (en vert) révélée au microscope électronique
Sources
Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice, Journal of Cell Biology, october, 15, 2012.
Iman Sahly (1,3,4), Eric Dufour (1,3,4), Cataldo Schietroma (1,3,4), Vincent Michel (2,3,4), Amel Bahloul (2,3,4), Isabelle Perfettini (2,3,4), Elise Pepermans (2,3,4), Amrit Estivalet (1,3,4), Diane Carette (2,3,4), Asadollah Aghaie (1,3,4), Inga Ebermann (2,3,4), Andrea Lelli (2,3,4), Maria Iribarne (2,3,4), Jean-Pierre Hardelin (2,3,4), Dominique Weil (2,3,4), José-Alain Sahel (1,5), Aziz El-Amraoui (2,3,4), and Christine Petit (1,2,3,4,6)
(1) Institut de la Vision, Syndrome de Usher et autres Atteintes Rétino-Cochléaires, Paris, France
(2) Institut Pasteur, Unité de Génétique et Physiologie de l'Audition, Paris, France
(3) Inserm UMRS587, Paris, France
(4) Inserm UPMC, Paris 6, France
(5) Inserm UMRS968, Institut de la Vision, Département de Génétique, Paris, France
(6) Collège de France, Paris, France
Contact presse
Service de presse de l'Institut Pasteur
Nadine Peyrolo / + 33 (0)1 45 68 81 46
Jérémy Lescène / +33 (0)1 45 68 81 01
presse@pasteur.fr


