

Researchers from the Institut Pasteur, the Institut de la Vision, Inserm, and the Pierre and Marie Curie University have determined the cause of blindness associated with type 1 Usher syndrome (the most common genetic cause of deafness and blindness in humans). They have also shown why rodents, the only animal model currently available for research on this disease, are invulnerable to the vision loss observed in human patients. Their work will be the basis for future research towards producing an animal model in primates. If successful, this could lead to a therapeutic approach for treating blindness in patients with type 1 Usher syndrome. This research was published on October 8th in the Journal of Cell Biology.
Press release
Paris, november 17, 2012
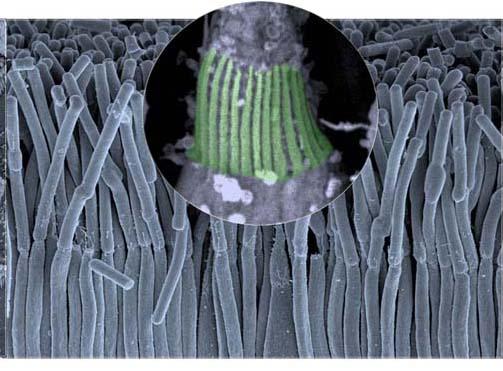
The work done by the team of Professor Christine Petit (2), head of the Genetics and Physiology of Hearing Unit (Institut Pasteur), in collaboration with Dr. Aziz El-Amraoui (Institut Pasteur) and Professor José-Alain Sahel (Institut de la Vision), has brought new hope as the researchers have recently discovered the cause of retinitis pigmentosa in patients suffering from type 1 Usher syndrome. It stems from a defect in the organization of cellular structures, called calyceal processes, necessary to maintain eyesight. This defect is caused by the dysfunction of one or more proteins, five of which were identified by the researchers, and which ensure the proper cohesion of calyceal processes. Researchers were able to observe the structure of these calyceal processes using high-resolution electron microscopy techniques (see image).
In their study, researchers also showed a major difference between rodent photoreceptor cells and those in primates. While primate photoreceptor cells include calyceal processes, rodent photoreceptor cells do not. This is why retinitis pigmentosa is not observed when the genes responsible for Usher syndrome in humans are mutated in mice.
Pioneers in the study of hereditary deafness, the team under Professor Christine Petit has identified three out of the five proteins that cause auditory impairments in type 1 Usher syndrome. Their research has most notably paved the way towards a better understanding of the molecular foundations of hearing. With these new findings, researchers hope to develop an improved animal model that will enable them to quickly put in place a therapeutic approach to treat retinitis pigmentosa in patients with Usher syndrome.
This research was supported by the European Community HEALTH-F2-2010-242013 (TREATRUSH), LHW-Stiftung, the Fondation Raymonde & Guy Strittmatter, FAUN Stiftung (Suchert Fondation), the Conny Maeva Chartiable Foundation, Fondation Orange, the French National Research Agency [ANR-10-LABX-65], [ANR-07-MRARE-009-01], the “Foundation Fighting Blindness Paris Center Grant", and the Fondation Voir et Entendre.
--
(1) Retinitis pigmentosa is found in a variety of hereditary retinal impairments characterized by the progressive degeneration of photoreceptor cells which leads to night blindness and a gradually reduced field of vision.
(2) Christine Petit is a professor at the Collège de France and the Institut Pasteur.
--
Illustration – Copyright Institut Pasteur
Caption : the structure of calyceal processes (green) seen under an electron microscope
Source
Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice, Journal of Cell Biology, October 15, 2012.
Iman Sahly (1,3,4), Eric Dufour (1,3,4), Cataldo Schietroma (1,3,4), Vincent Michel (2,3,4), Amel Bahloul (2,3,4), Isabelle Perfettini (2,3,4), Elise Pepermans (2,3,4), Amrit Estivalet (1,3,4), Diane Carette (2,3,4), Asadollah Aghaie (1,3,4), Inga Ebermann (2,3,4), Andrea Lelli (2,3,4), Maria Iribarne (2,3,4), Jean-Pierre Hardelin (2,3,4), Dominique Weil (2,3,4), José-Alain Sahel (1,5), Aziz El-Amraoui (2,3,4), and Christine Petit (1,2,3,4,6)
(1) Institut de la Vision, Syndrome de Usher et autres Atteintes Rétino-Cochléaires, Paris, France
(2) Institut Pasteur, Genetics and Physiology of Hearing Unit, Paris, France
(3) Inserm UMRS587, Paris, France
(4) Inserm UPMC, Paris 6, France
(5) Inserm UMRS968, Institut de la Vision, Département de Génétique, Paris, France
(6) Collège de France, Paris, France
Contacts
Institut Pasteur Press Office
Nadine Peyrolo – +33 (0)1 45 68 81 46
Jérémy Lescene – +33 (0)1 45 68 81 01
presse@pasteur.fr


